Blang
Blang
Tools for Bayesian data science and probabilistic exploration
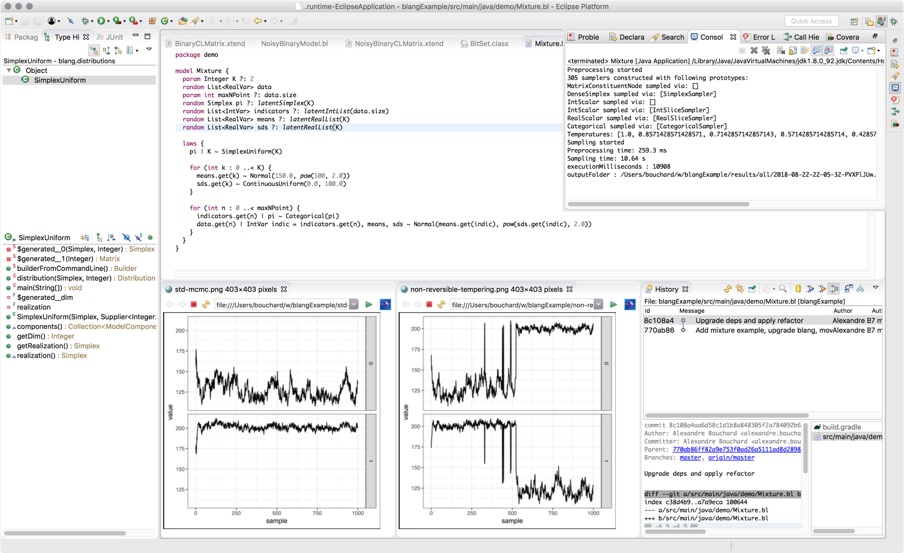
Blang is a language and software development kit for doing Bayesian analysis. Our design philosophy is centered around the day-to-day requirements of real world data science. We have also used Blang as a teaching tool, both for basic probability concepts and more advanced Bayesian modelling. Here is the one minute tour:
The above example illustrates several aspects of Blang:
-
Blang goes beyond simple real and integer valued random variables. Here we have a type of phylogenetic tree,
UnrootedTree. Bayesian inference over combinatorial spaces is important and currently neglected. So Blang uses an open type system and assists you in creating complex random types and correct sampling algorithms for these types. -
All the distributions shown, e.g.
Exponential,NonClockTreePrior, etc, in particular, those in the SDK, are themselves written in Blang. This is important for extensibility and, crucially, for teaching. When using the Blang IDE, students can command-click on a distribution to jump to its definition in the language they are familiar with. -
The parameters of the distributions can themselves be distributions, e.g.
NonClockTreePriortaking in aGammadistribution as argument. This is useful to create rich probability models such as Bayesian non-parametric priors. -
As hinted by the keyword
package, you can use other people's models, and package yours easily (and in a versioned fashion). This is useful to disseminate your work and critical to create reproducible analyses. Details such as dependency resolution are taken care of automatically.
If you have one more minute to spare, let us see what happen when we run this model (if you want to try at home, all you need to run this is Open or Oracle SDK 8 SDK and git installed):
-
We inferred a distribution over unobserved phylogenetic trees given an observed multiple sequence alignment.
-
The engine used here,
SCM(Sequential Change of Measure) is based on state-of-the-art sampling methods. It uses Jarzynski's method (with annealed distributions created automatically from the model) with a Sequential Monte Carlo algorithm and an adaptive temperature schedule. Other methods include Parallel Tempering, various non-reversible methods, and users can add other inference frameworks as well. -
The algorithm trivially parallelize to a large number of CPUs, here only 8 cores were used. Massive distribution over many nodes is in the pipeline.
-
The method provides an estimate of the evidence, here
-1216.56, which is critical for model selection and lacking in many existing tools. Running again the method with twice as many particles, we get-1216.00, suggesting the estimate is getting close to the true value. In contrast, variational methods will typically only give a bound on the true evidence. -
The resulting samples are easy to use: tidy csv's in a unique execution folder created for each run. You can integrate Blang in your data analysis pipeline seamlessly.
-
The command line arguments are automatically inferred from the random variables declared in the model and the constructors of the corresponding types (with a bit of help of some annotations).
-
Blang is built using Xtext, a powerful framework for designing programming languages. Thanks to this infrastructure, Blang incorporates a feature set comparable to many modern full fledge multi-paradigm language: functional, generic and object programming, static typing, just-in-time compilation, garbage collection, IDE support leverage static types and including debugging, etc.
-
Blang runs on the JVM, so it is reasonably fast (typically within a small factor to any contender) without resorting to rewriting inner loops into low-level, error prone languages. You can also call any Java or Xtend code, and there is a good interoperability potential with the industrial data science stack such as Hadoop, Spark and DL4J.
-
Blang is free and open source (permissive Berkeley License for both the SDK and the language infrastructure).