Alexandre Bouchard-Côté

Professor of Statistics
University of British Columbia
bouchard@stat.ubc.ca
Research highlights
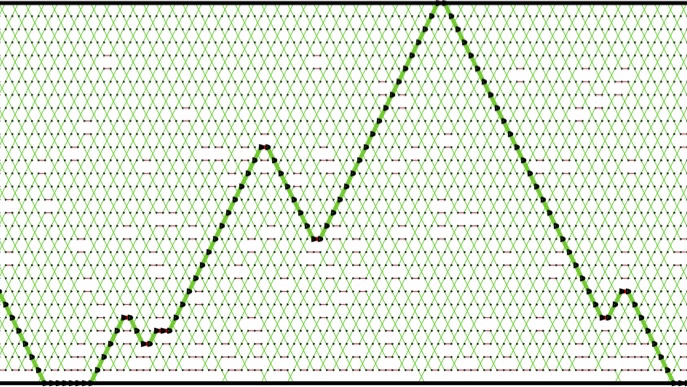
Scalable approximation of complex probability distributions
The prevalence of uncertainty in our world has fuelled the development of sophisticated mathematical methods to understand and tame uncertainty. This has been a central quest in the field of statistics. A key concept often used to depict uncertainty is the notion of a probability distribution, which can be thought of as measuring, for each possible state of the system, a degree of belief. Being able to interrogate probability distributions is therefore of paramount importance in statistics, and hence in the many fields of science and engineering that depend on statistics and uncertainty quantification. As scientific models become increasingly complex, the calculations required to query probability distributions are getting computationally prohibitive, to the point that these computations are the bottleneck in many disciplines. My field of research is concerned with computational methods that break these bottlenecks, by making use of algorithms exploiting randomness.
JRSSB paper on a new perspective to Parallel Tempering (PT)
ICML paper on PT with generalized paths of distributions
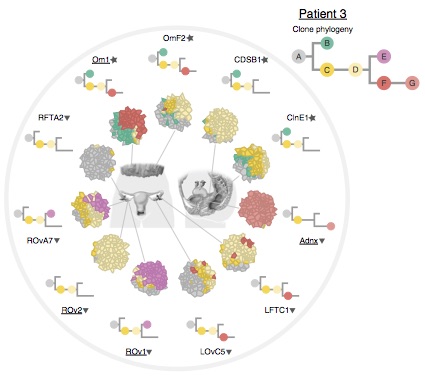
Probabilistic modelling of the evolutionary dynamics and phylogeny of cancer
Proliferating cancer cells, in which DNA repair mechanisms are disrupted, accumulate mutations at a much faster rate than healthy cells do. This leads to the emergence of an evolutionary process inside the tumour. A current research frontier is the characterization of the evolutionary dynamics and phylogenies within individual cancer tumours, where multiple sub-populations of cancer cells acquire differentiating sets of mutations.
Phylogenetic inference from single cell copy number alterations
Nature Methods paper on the analysis of single cell data
Nature Methods paper on PyClone, a Bayesian non-parametric deconvolution method for bulk cancer data

Tools for Bayesian data science
We are developing a language and software development kit for doing Bayesian analysis. The design philosophy is centered around the day-to-day requirements of real world (Bayesian) data science. The inference engines brings to bear several recent advances such as non-reversible methods.
Easy to use distributed Bayesian inference
A modelling language for Bayesian inference over combinatorial spaces
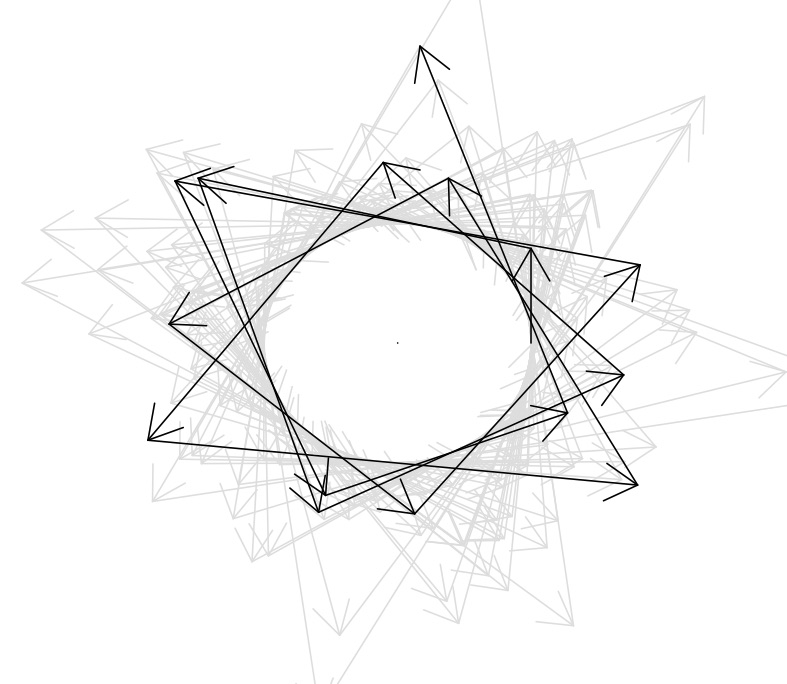
Non-reversible gradient-based Monte Carlo methods
Markov chain Monte Carlo (MCMC) is notoriously difficult to scale to problems having high-dimensional latent variables ("big models"), which arise in many scientific and engineering applications.
We are working on an alternative to MCMC that we call the "Bouncy Particle Sampler" (BPS), which imports ideas from the field of molecular simulation to scale MCMC to high dimensional problems. JASA paper
Follow-up Annals of Statistics paper, on the geometric ergodicity of BPS
Preprint of follow-up work, on non-linear trajectories and discrete piecewise deterministic Markov processes
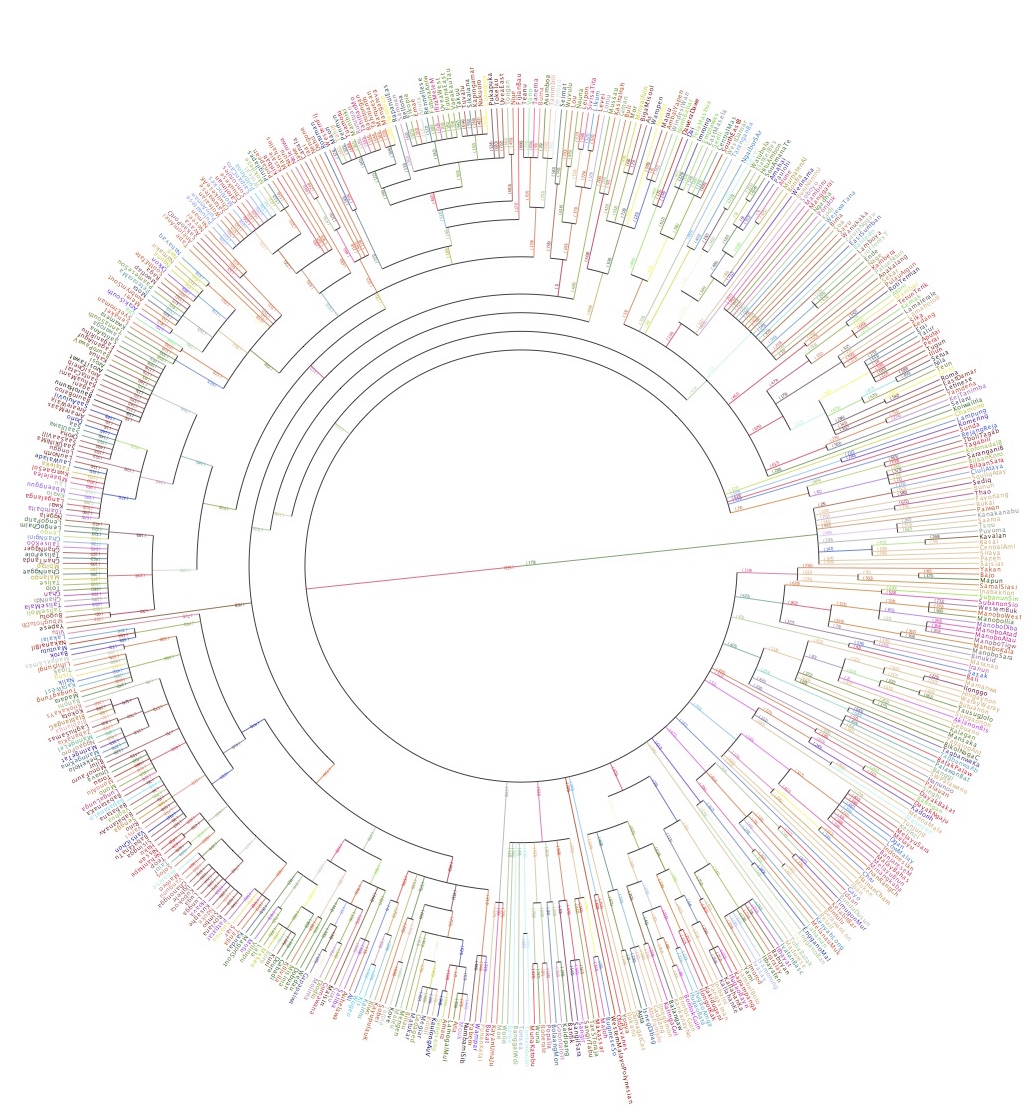
Bayesian phylogenetic inference
As a result of advances in sequencing technologies, the fields of computational and statistical phylogenetics, which are concerned with the modelling and inference of evolutionary relationships, have been growing rapidly in recent years. I am particularly interested in computationally-intensive Bayesian methods and inference of complex evolutionary models.
Sys Bio paper on change-of-measure based phylogenetic SMC algorithm.
Novel sampling method based on Hamiltonian Monte Carlo for parameter-rich evolutionary models.
Long indel model (in Sys Bio) based on the Poisson Indel Process (PNAS).
Computational historical linguistics
Phylogenetic trees (or networks, forests, etc) also play an important in linguistics, to describe how language change and splits in ancestral speaker populations gave rise to today's linguistic diversity. Computational methods are also starting to play an important role in this field.
Bio
My main field of research is computational statistics. I am interested in the mathematical side of the subject as well as in applications in astronomy, biology and linguistics.
On the methodology side, I am interested in Monte Carlo methods such as SMC and MCMC, probabilistic modelling (including Bayesian statistics), probabilistic programming and variational inference.
A lot of my applied work involves difficult integration problems, motivating my choice of methodological topics. In particular, I am interested in "computational Lebesgue integration", including integration over mixed combinatorial spaces (in, e.g, cancer phylogenetics, tree and alignment inference, automated reconstruction of proto-languages, population genetics, etc).
Academic background
- University of California, Berkeley (2005–2010).
- PhD in Computer Science with a Designated Emphasis in Statistics.
- Minor in Linguistics.
- Research advisors: Michael I. Jordan and Dan Klein.
- Title of dissertation: Probabilistic Models of Evolution and Language Change.
- McGill University (2002–2005).
- Joint Honours Program in Mathematics and Computer Science.
- Research advisors: Doina Precup and Prakash Panangaden.
Employment
- University of British Columbia (2011–Present).
- Professor of Statistics (2022–Present)
- Associate Professor of Statistics (2016–2022)
- Assistant Professor of Statistics (2011–2016)
Awards
- CRM-SSC Prize in Statistics (2024)
- PIMS-UBC Mathematical Sciences Young Faculty Award (2018)
- Tweedie New Researcher Award, Institute of Mathematical Statistics (2016)
- Google Faculty Award (2013)
- Martha Piper Award (2012)
- NeurIPS Best Student Paper Award (2011)
- Siebel Scholar (2010)
- Bourse du Fond de Recherche du Québec (2010)
- NSERC Post-Graduate Scholarship (2008)
- Courtemanche Scholarship (2004)
- James McGill Scholarship (2002)